NHS screening programme for sickle cell and thalassaemia
NHS Screening Programme for Sickle Cell and Thalassaemia
Our goal is to develop a linked programme of high quality screening and care in order to:
- support people to make informed choices during pregnancy and before conception
- improve infant health through prompt identification of affected babies
- provide high quality and accessible care throughout England
- promote greater understanding and awareness of the disorders and the value of screening
Key messages from UK National Screening Committee:
- Offer screening pre-conceptually
- Offer screeningas early as possible, but at least by 8-10 weeks of pregnancy
- Offer all pregnant women screening for sickle cell and thalassaemia
- Observing local policy, base screening on the Family Origin Questionnaire and blood test results
- All babies can be screened for sickle cell disorders on the newborn bloodspot
- http://sct.screening.nhs.uk/
Sickle Cell and Thalassaemia
- Sickle Cell Disorders
- autosomal recessively inherited. Sickle Cell Disorders (which include HbSS, HbSC, HbSD Punjab, HbSb Thal (b+,b0, db, Lepore), HbSO Arab, Hb-S/HPFH) are a variable set of conditions which can cause chronic anaemia, jaundice, painful crisis, organ damage, infections and strokes.
- there are approximately 240,000 healthy carriers and more than 14,500 people with Sickle Cell Disorders in the UK. All pregnant women with Sickle Cell Disorders should receive specialist obstetric and haematological care
- Thalassaemia
- Alpha and Beta Thalassaemia Major are autosomal recessively inherited
- are approximately 214,000 healthy carriers and more than 700 people with Thalassaemia Disorders in the UK
- Alpha Thalassaemia Major is incompatible with intrauterine life
- Beta Thalassaemia Major results in life threatening anaemia and requires blood transfusions every 4-6 weeks and iron chelation to prevent further illness.
- all pregnant women with Thalassaemia Disorders should receive specialist obstetric and haematological care
- screening policy for sickle cell and thalassaemia
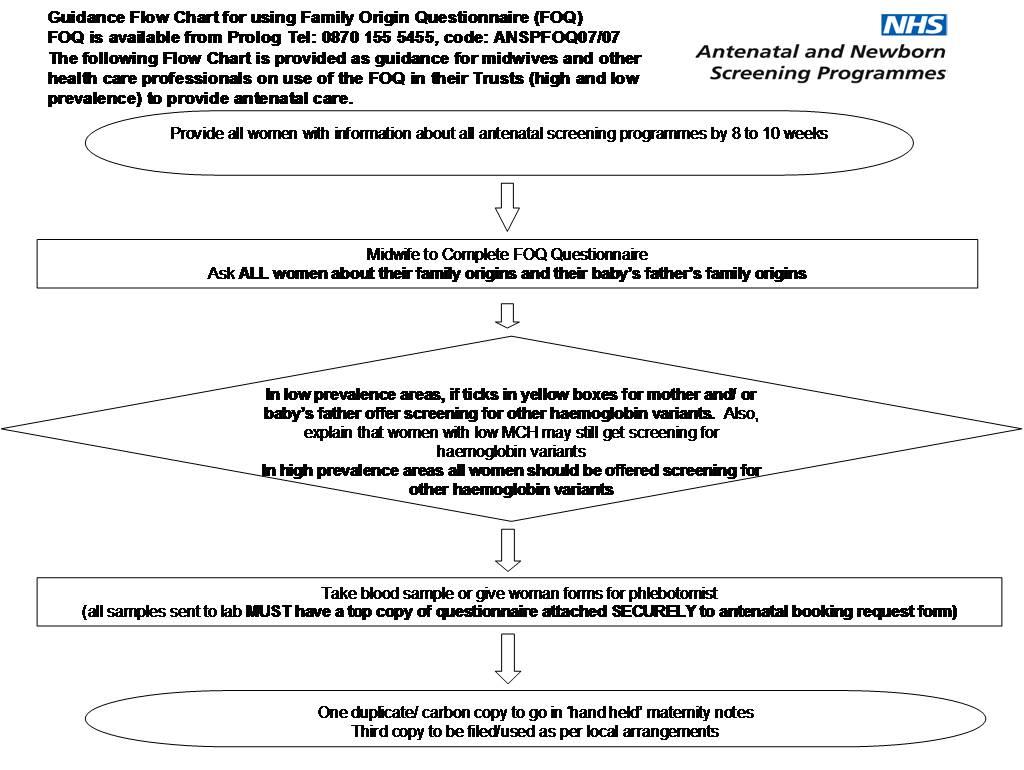
- Screening Policy Screening for Sickle Cell and Thalassaemia and other haemoglobin variants in pregnancy should be performed early, preferably between 8 -10 weeks.
- there are two approaches to the offer of screening depending on whether a Trust is defined as either high or low prevalence
- high prevalence trusts are those where sickle cell diseases are estimated to affect over 1.5 pregnancies per 10,000 births. Information from the FOQ is used to interpret the blood results
- low prevalence trusts are those where sickle cell diseases are estimated to affect less than 1.5 pregnancies per 10,000 births. The FOQ is used to screen women and the baby’s father and assess the risk of haemoglobin variants and sickle cell disease
- Thalassaemia screening using routine blood indices will be offered to all women in England, i.e. in high and low prevalence Trusts. Women should be aware of this and informed consent should be sought prior to screening
- screening for sickle cell and other haemoglobin variants will depend on the prevalence of the condition. Sickle cell and other haemoglobin variants screening is offered to all women in those Trusts identified as high prevalence. All other areas will be required to offer, as a minimum, laboratory testing for sickle cell and other haemoglobin variants based on an assessment of risk determined by a question to women about their and their baby's father's family origin. Copies of the family origin questionnaire are available from the following website:
- there are two approaches to the offer of screening depending on whether a Trust is defined as either high or low prevalence
- Screening Policy Screening for Sickle Cell and Thalassaemia and other haemoglobin variants in pregnancy should be performed early, preferably between 8 -10 weeks.
- professional Education for Genetic Assessment and Screening training resources are available at http://www.pegasus.nhs.uk
- two reports which have informed policy and standards are:
- HTA 1999; Vol 3: No 11 Antenatal and neonatal Haemoglobinopathy screening in the UK: Review and economic analysis
- HTA 2000; Vol4:No 3 Screening for sickle cell and thalassaemia: a systematic review with supplementary research
- NICE have suggested (1)
- screening for sickle cell diseases and thalassaemias should be offered to all women as early as possible in pregnancy (ideally by 10 weeks). The type of screening depends upon the prevalence and can be carried out in either primary or secondary care
- where prevalence of sickle cell disease is low (fetal prevalence 1.5 cases per 10,000 pregnancies or below), all pregnant women should be offered screening for haemoglobinopathies using the Family Origin Questionnaire:
- if the Family Origin Questionnaire indicates a high risk of sickle cell disorders, laboratory screening (preferably high-performance liquid chromatography) should be offered
- if the mean corpuscular haemoglobin is below 27 picograms, laboratory screening (preferably high-performance liquid chromatography) should be offered
- if the Family Origin Questionnaire indicates a high risk of sickle cell disorders, laboratory screening (preferably high-performance liquid chromatography) should be offered
- screening for sickle cell diseases and thalassaemias should be offered to all women as early as possible in pregnancy (ideally by 10 weeks). The type of screening depends upon the prevalence and can be carried out in either primary or secondary care
For more information then see http://www.screening.nhs.uk/
Reference
- NICE. Antenatal care. NICE guideline NG201. Published August 2021
Related pages
- Antenatal and newborn screening
- Thalassaemia
- Sickle cell anaemia
- Family origin questionnaire (FOQ) - recessive inheritance tool
- Diagram of inheritance pattern of an autosomal recessive condition e.g. Sickle Cell Disease, Thalassaemia, Cystic Fibrosis
- Diagram of parental carrier state combinations that give rise to the risk of a fetus with significant sickle cell disease or beta thalassaemia
- NHS screening programme patient information leaflets for sickle cell disease and thalassaemia
- NICE - pregnancy screening guidance for thalassaemia and sickle cell disease
Create an account to add page annotations
Add information to this page that would be handy to have on hand during a consultation, such as a web address or phone number. This information will always be displayed when you visit this page