Bilateral acoustic neurofibromatosis, also referred to as Type 2, NF2 or central neurofibromatosis, is clinically and genetically distinct from NF1. NF2 involves tumours of the vestibular branch of the eighth cranial nerve, namely vestibular schwannomas.
- billateral vestibular schwannomas (VS) are emblematic of NF2, but schwannomas may form on other cranial, spinal, and peripheral nerves
- central nervous system meningiomas and ependymomas, peripheral neuropathy, amyotrophy, retinal hamartomas, and subcapsular lens opacities are part of the NF2 disease spectrum (1,2, 3).
Patients with NF2 may also have peripheral neurofibromas, cafe-au-lait spots, and meningiomas. The NF2 gene is an example of a tumour suppressor gene. It is located on the long arm of chromosome 22 and is inherited as an autosomal dominant trait
- somatic mosaicism is present in about one-third of de novo patients (1, 2)
- germline truncating mutations are associated with more severe disease than large deletions and missense mutations; individuals with germline truncating mutations are diagnosed at a younger age and usually have an earlier onset of symptomatic tumours
NF2 is an inherited tumour suppressor disease with a prevalence of 1 in 60,000 and a birth incidence of 1 in 25-30,000 individuals (1)
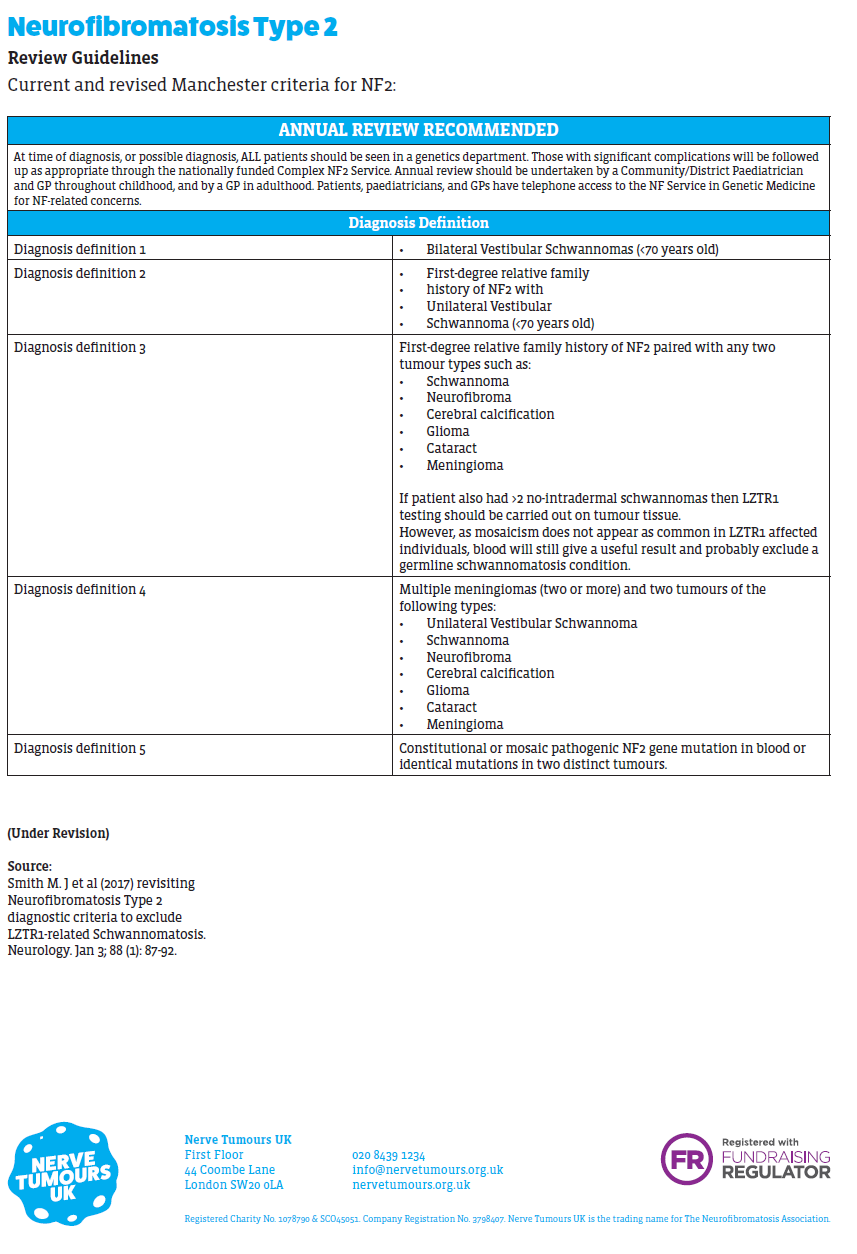
Reference:
- Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
- Evans DGR, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F. Birth incidence and prevalence of tumour prone syndromes: estimates from a UK genetic family register service. Am J Med Genet A. 2010;152A(2):327-332.
- Ferner R. Neurofibromatosis 1 and neurofibromatosis 2: a twenty-first century perspective. Lancet Neurol. 2007;6(4):340-345.
Related pages
Create an account to add page annotations
Add information to this page that would be handy to have on hand during a consultation, such as a web address or phone number. This information will always be displayed when you visit this page