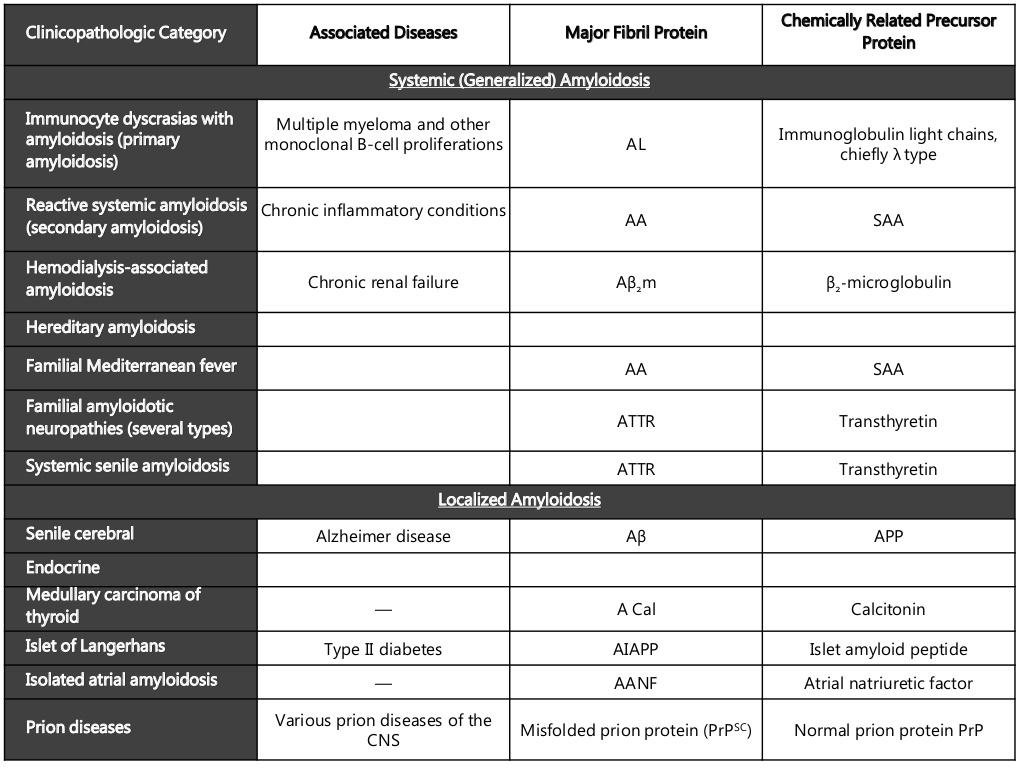
Verschiedene Proteine können als Amyloid in verschiedenen Geweben abgelagert werden
- Ablagerungen können auf eine bestimmte Stelle beschränkt sein (z. B. Bauchspeicheldrüse, Gehirn, Kehlkopf) (lokalisierte Amyloidose) oder im ganzen Körper vorkommen (systemische Amyloidose)
- es gibt viele Arten der lokalisierten Amyloidose, einschließlich der zerebralen endokrinen und der kutanen Amyloidose
- nur der Name Amyloid ist identisch, aber die oben erwähnten lokalisierten Amyloidformen sind aufgrund unterschiedlicher Vorläuferproteine völlig verschieden
- nur der Name Amyloid ist identisch, aber die oben erwähnten lokalisierten Amyloidformen sind aufgrund unterschiedlicher Vorläuferproteine völlig verschieden
- es gibt viele Arten der lokalisierten Amyloidose, einschließlich der zerebralen endokrinen und der kutanen Amyloidose
- Die fünf Haupttypen der systemischen Amyloidose sind:
- Die systemischen Formen von Amyloid, die voneinander unterschieden werden sollten (vier erworbene und eine erbliche), sind AA, AL, Aß2M und zwei ATTR-Typen:
- erworbene AA-Amyloidose
- verursacht durch chronische Entzündungen
- Das Serum-Amyloid-A-Protein (SAA), ein Akute-Phase-Reaktant, ist das Vorläuferprotein dieses Typs
- Proteinurie und Verlust der Nierenfunktion sind die wichtigsten klinischen Merkmale
- erworbene AL-Amyloidose
- verursacht durch eine Plasmazelldyskrasie
- Vorläuferprotein dieses Typs ist eine leichte Kappa- oder Lambda-Immunglobulin-Kette
- Die klinischen Merkmale dieses Typs sind sehr vielfältig, z. B. Kardiomyopathie, Hepatomegalie, nephrotisches Syndrom, schwere Diarrhö, Karpaltunnelsyndrom und Neuropathie (periphere und autonome Neuropathie)
- erworbene Aß2M-Amyloidose
- verursacht durch chronische Dialyse aufgrund eines vollständigen Nierenversagens
- Vorläuferprotein dieses Typs ist das ß2-Mikroglobulin, da die hohen Serumspiegel das Ergebnis der Unfähigkeit sind, dieses Protein auszuscheiden
- Klinische Merkmale dieses Typs sind Karpaltunnelsyndrom und Gelenkprobleme (Schultern, Handgelenke, Finger, Hüften, Wirbelsäule usw.)
- verursacht durch chronische Dialyse aufgrund eines vollständigen Nierenversagens
- erworbene ATTR-Amyloidose
- tritt im höheren Lebensalter auf (insbesondere über 80 Jahre)
- das normale Wildtyp-Vorläuferprotein Transthyretin (TTR) ist das charakteristische Protein
- klinisch gekennzeichnet durch eine langsam fortschreitende Kardiomyopathie
- hereditäre ATTR-Amyloidose
- verursacht durch mehr als 80 autosomal dominant vererbte Punktmutationen des Vorläuferproteins Transthyretin (TTR)
- Klinische Merkmale dieses Typs sind periphere und autonome Neuropathie, aber auch Herz-, Nieren- und Augenbeteiligung können vorkommen
- erworbene AA-Amyloidose
- Die systemischen Formen von Amyloid, die voneinander unterschieden werden sollten (vier erworbene und eine erbliche), sind AA, AL, Aß2M und zwei ATTR-Typen:
Referenz:
- Misumi Y, Ando Y. Klassifizierung der Amyloidose. Brain Nerve. 2014 Jul;66(7):731-7.
- Dey A. Classification of Amyloidosis.
Verwandte Seiten
Erstellen Sie ein Konto, um Seitenanmerkungen hinzuzufügen
Fügen Sie dieser Seite Informationen hinzu, die Sie während eines Beratungsgesprächs benötigen, z. B. eine Internetadresse oder eine Telefonnummer. Diese Informationen werden immer angezeigt, wenn Sie diese Seite besuchen